

Abstract
Introduction
Multiple myeloma (MM) is a result of a malignant transformation of plasma cells that is preceded by the presence of an asymptomatic clonal plasma cell expansion, a condition referred to as monoclonal gammopathy of undetermined significance (MGUS). We and others have shown familial aggregation of MM and MGUS. Evidence from epidemiologic, family and genome-wide association studies (GWAS) suggests a genetic component underlying MM etiology. GWAS have successfully established 17 common genetic risk loci for MM to date and recently, rare inherited susceptibility variants in the LSD1 / KDM1A and USP45 genes were identified in familial MM / MGUS kindreds. Family-based approaches may be used to elucidate genetic variation contributing to familial MM. Genetic linkage analysis has historically been used to detect the chromosomal location of disease genes. The objective of this study was to conduct a linkage analysis of MM / MGUS families to identify genomic regions for MM / MGUS.
Methods
Linkage analysis was performed on whole exome sequencing (WES) data generated from germline DNA extracted from peripheral blood from MM / MGUS families ascertained at four sites, of which 79 were selected with 2 or more affected members with any combination of MM or MGUS. Whole exome capture was performed using Agilent SureSelect 38 Mb paired end sequencing and ran on Illumina HiSeq 2000s/2500s; standard analyses for alignment (GRCh37) and quality control were conducted. The Genome Analysis Toolkit's (GATK) HaplotypeCaller in per-sample mode was used to jointly call all the samples together. Quality control included removing variants that had <75% call rate, <8x coverage, or minor allele frequency <0.01 (1KGenomes). The jointly called WES data from the 79 families consisted of 230 individuals, including 119 MM, 93 MGUS and 18 unaffected relatives. Additional early onset (defined as age <60) MM cases (n=400), sporadic MM cases (n=879) and controls (n=2389) were also included in the joint calling to be used for follow-up of linkage regions. We included independent variants defined as linkage disequilibrium (LD) r2 <0.05 using both PLINK 1.07's LD based variant pruner and also MERLIN's pairwise r2 cluster marker-marker modeling to ensure variant independence. After quality control and LD filtering: 12,946 variants remained. We conducted multipoint linkage analysis using MERLIN (Multipoint Engine for Rapid Likelihood Inference using the Lander-Green approach) and considered both non-parametric and parametric (dominant and recessive) models to test for co-segregation of chromosomal regions with MM / MGUS. A logarithm of the odds (LOD) score greater than 3.3 was significant evidence for linkage. Within the same families, we interrogated any suggestive regions by performing gene-level association tests using the PEDGENE R package to calculate burden and kernel statistics, accounting for family relationships. Follow-up of these genes in the early onset and sporadic MM cases and controls and interrogation of single variants driving the associations of these genes is ongoing.
Results
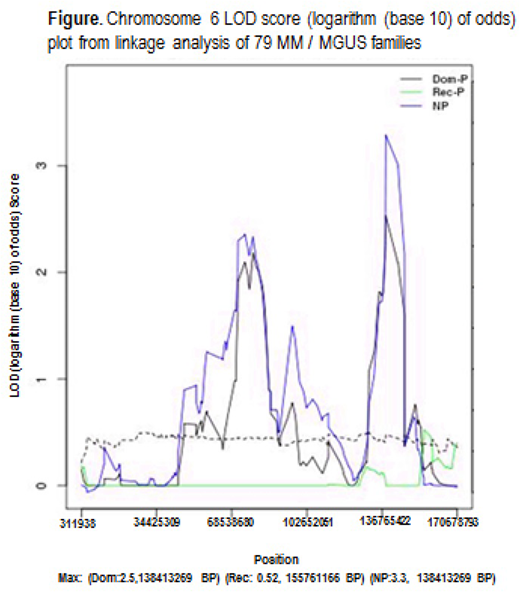
Among the 79 families, 28 consisted of two or more MM, 41 had two or more MM and MGUS cases and 10 had two or more MGUS. Analyses of the 79 MM / MGUS families identified chromosome 6 at 123420001-149070000 BP region, with evidence for linkage by the non-parametric model (LOD score=3.3) and supportive evidence for linkage by the dominant parametric model (LOD=2.5) (Figure). The chromosome 6 region, which is outside the HLA-region, contains a total of 72 genes. Using gene-level association tests, we identified four genes within the region (MOXD1, TRDN, ADAT2, LTV1) that were associated within MM / MGUS in the family cases (p<0.05).
Discussion
We found evidence for a locus on chromosome 6 linked to MM / MGUS. Four genes in this region may be associated with MM / MGUS in these families. Follow-up of these genes/variants in high-risk individuals (early-onset / age <60), sporadic cases and controls will help elucidate genetic mechanisms contributing to familial MM / MGUS. In addition, we demonstrate the benefit of applying a linkage analysis framework to familial WES data for discovery of genomic regions influencing MM / MGUS.
Dumontet:Roche: Research Funding; Merck: Consultancy, Membership on an entity's Board of Directors or advisory committees; Janssen: Honoraria; Sanofi: Honoraria. Kumar:AbbVie: Membership on an entity's Board of Directors or advisory committees, Research Funding; Janssen: Membership on an entity's Board of Directors or advisory committees, Research Funding; Celgene: Membership on an entity's Board of Directors or advisory committees, Research Funding; AbbVie: Membership on an entity's Board of Directors or advisory committees, Research Funding; Oncopeptides: Membership on an entity's Board of Directors or advisory committees; Takeda: Membership on an entity's Board of Directors or advisory committees; Merck: Membership on an entity's Board of Directors or advisory committees, Research Funding; Celgene: Membership on an entity's Board of Directors or advisory committees, Research Funding; KITE: Membership on an entity's Board of Directors or advisory committees, Research Funding; Novartis: Research Funding; Roche: Research Funding; Janssen: Membership on an entity's Board of Directors or advisory committees, Research Funding; KITE: Membership on an entity's Board of Directors or advisory committees, Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal